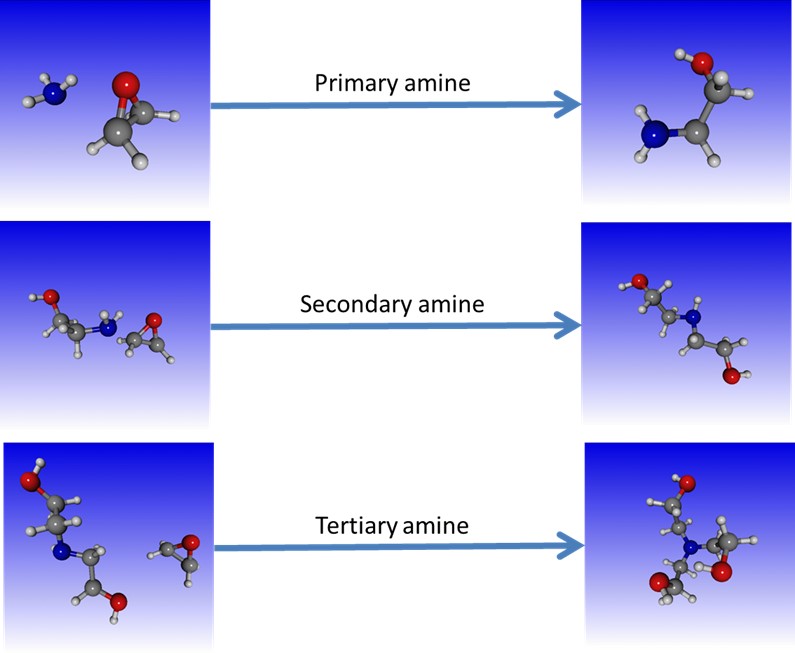
Epoxy is undoubtedly one of the most successful matrix in the field of polymer composites. It attracts a lot of attention from researchers and industrials with the common objective to formulate a stiff, strong, and tough resin. Such a goal can only be reached if the fundamental mechanisms which control the final properties of the resin are well understood and controlled. The nanoscale simulation techniques nowadays become effective approaches to complement high-tech experiments and offer an alternative route to understanding the properties of complex systems. Among the various techniques developed over the past half-century, molecular dynamics has gained popularity thanks to both its simple principle and its efficiency in tackling versatile problems. However the key parameter of the simulation, the force field which models the interactions between the atoms, is traditionally classified as non-reactive, i.e. it cannot model the formation and disappearance of covalent bonds between functional chemical moieties. Modelling the curing process of an epoxy system remains therefore highly challenging. To circumvent this issue, the ReaxFF reactive force field is used for the first time to predict the reactivity of epoxy precursor and hardener molecules. It is capable of modelling non-catalytic reactions between epoxide and amine groups as well as autocatalytic reactions between hydroxyl and amine groups. The formation of the 3D network is monitored by following either the number and mass of molecules or the number of reactive groups in the system. This original modelling work does not only mimic real systems but also gives a molecular view of the mechanisms which drive the curing process. This study is a first step toward the development of a multi-scale modelling framework aiming at giving a comprehensive view of the physical chemistry of nano-engineered fiber reinforced composites.